

Non-linear pharmacokinetics is a change of pharmacokinetic process from primarily first order to zero order with increasing size of dose administration. Sometimes it is a mixture of the first order and zero order that, ‘s why it is called mixed order kinetics. From linear pharmacokinetics, the deviation is seen that’s why it is also called non-linear pharmacokinetics. Primarily, the Dose of administration of the drug is directly proportional to the ADME of the drug then it is said to be first order or linear kinetics.
The rate process of ADME is susceptible to high drug concentration. In such an instant, zero-order kinetic parameters change depending on the quantity of dosage form. Non-linear pharmacokinetics can be known as dose-dependent pharmacokinetics.
Test to detect nonlinearity in pharmacokinetics:
- Determination of important parameters such as elimination half-life, fraction bioavailable, or total systemic clearance. It is termed as the second test.
- Determination of steady-state plasma concentration at different doses. It is termed as the first test.
Eg: Glycine conjugation of salicylate, Sulphate conjugation of salicylamide, Elimination of Phenytoin
Michaelis-menton method of estimating parameters:
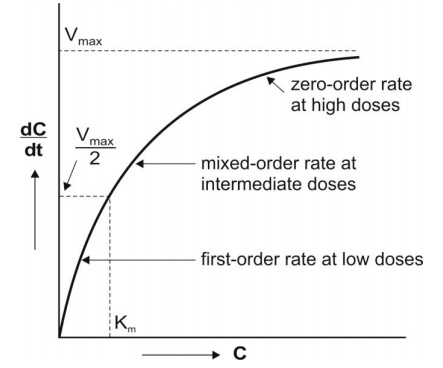
Michaelis-mention equation is a method that describes the kinetics of capacity limited. It explains the difference in the rate of the chemical reaction catalyzed by enzymes. It is to understand enzyme kinetics It is expressed by the following:

Where,
- –dC/dt: rate of decline of drug concentration with time,
- Vmax = theoretical maximum rate of the process
- Km = Michaelis constant, substrate conc at which rate of reaction is half of the maximum velocity.
According to the equation, three situations can be considered:
1. Where, KM= C
Then, -dC/dt = Vmax / 2
2. If a drug at low conc. KM >> C:
Here, KM + C = KM, And the Michaelis-menton equation become
-dc/ dt = VmaxC/ Km This equation is similar to the equation that shows the first order.
3. If, KM << C :
Then the Michaelis- monton equation becomes, -dc/ dt = Vmax
The above equation is identical to that described in Zero order.
Practically, graphically Kmax and Vmax compute in 3 ways:
1. Lineweaver- Burk plot:
At a steady state, the rate of dosing equals to rate of decline in plasma drug conc and if the decline is due to a capacity-limited process, then:
DR= Vmax Css/ Km + Css
If we take the reciprocal of the above equation we get
1/DR = Km/ Vmax Css + 1/ Vmax The plot of 1/ DR vs 1/ Css yields a straight line with a slop Km/ Vmax and 1/ Vmax of the y-intercept.
2. Direct Linear Plot:
A pair of Css 1 and Css 2 obtain dosing rated DR 1 and DR 2 respectively. Joining points Css1 and DR1 form a line, and the second line is obtained by joining points Css 2 and DR 2, when these two are drawn and see the intersecting point on Yaxis Vmax is understood and on X-axis Km is understood.
3. Graphical method:
If we rearrange equation, DR= Vmax Css/ Km + Css we get DR= Vmax – Km DR/ Css’ eq 1
It is observed that K m is much less variable than Vmax.
Limitations of Kmax and Vmax by assuming a compartment system, Kmax and Vmax will be larger when:
- If the drug is eliminated by more than one process.
- If the drug exhibits a first-order elimination process.
- If the drug follows multiple kinetics.
Causes of non-linearity pharmacokinetics:
- Saturation of enzymes in the process of drug ADME
- Pathological alteration of ADME of the drug
- The binding site of plasma protein or tissue gets saturated.
- Non-linearity in pharmacokinetics caused due to capacities of limited metabolism.
- Saturation of enzyme
- Saturation of active tubular secretion and reabsorption.
Factors causing nonlinearity:
The various factors causing nonlinearity are discussed below:
Drug absorption:
Non-linearity in drug absorption is caused by 3 important sources
- When absorption is dissolution or solubility rate is limited:
- When absorption is through carrier-mediated transport systems.
- When hepatic metabolism attends saturation.
Gastric emptying and GI blood flow are also causes of the non-linearity of drug absorption.
Drug distribution:
In distribution nonlinearity of drugs administered at high doses may be due to:
- Saturation of tissue binding.
- Saturation of binding sites on plasma protein.
In these two cases, With increasing plasma drug conc only Vd increases. Clearance also changes depending on the extraction ratio of the drug. The pharmacologic response can increase. Clearance which is unbound with low ER is unaffected and increases in pharmacologic response.
Drug metabolism:
Small changes in dose administration can affect the steady state of plasma concentration therefore, nonlinear kinetics is capacity-limited metabolism. It also demonstrates saturation limited metabolism in humans.
Important causes of non-linear metabolism are:
- Saturation of cofactor and/ or enzyme cause of capacity-limited metabolism
- Induction of enzyme eg. Carbamazepine. Enzyme induction is a common cause of both time and dose-dependent kinetics.
Drug excretion:
It is caused by saturation of renal excretion:
- Active tubular reabsorption eg. Glucose
- Active tubular secretion eg. Penicillin G
Active processes such as biliary secretion can also saturate. Eg Indomethacin.
Examples of drugs showing Non-linear pharmacokinetics:
GI Absorption :
- Intestinal metabolism eg, Riboflavin, Ceftibuten and baclofen.
- Saturable transport in gut wall eg, Propranolol, Salicylamide.
- Saturable transport in gut wall eg, Propranolol, Salicylamide.
Distribution:
- Cellular uptake eg, Methicillin.
- CSF transports eg, Benzylpenicillin
- Tissue binding eg, Imipramine
- Saturable transport into or out of tissue eg, Methotrexate.
Renal elimination:
- Active secretion eg, Mezlocillin
- Changed in urine eg, Salicylic acid
- Tubular reabsorption eg, Ascorbic acid
Metabolism
- Enzyme induction eg, Carmazipine.
- Saturated metabolism eg, Phenytoin, Salicylic acid
- Altered hepatic blood flow eg, Verpamil, Propranolol
- Metabolic inhibition eg, Diazepam
- Cofactor and enzyme limitation eg, alcohol, and acetaminophen.
Biliary Excretion
- Enterohepatic recycling eg, Cimetidine
- Billary secretion eg, Iodipamide
Non-linear pharmacokinetic, Phenytoin:
Phenytoin is a drug that is known to have a Km value before the therapeutic range. Therefore patients can overdose by drug administration. At low conc conc of half-life is 12 hrs, whereas in higher concentration it may be 24 hours.
The dose of administration of this drug every 12 hours can be lethal. The concentration of a drug above 20mg/L elimination may be low. Typical Vm values are 300 to 700 mg/ day. Which can be eliminated by these patients per day. This drug can accumulate in the body which can be dangerous.
References:
- DM Bramhandkar Sunil B. Jaiswal, Biopharmaceics and pharmacikientics, pg no 273- 281.




